
SANDWICH Elisa
Principle:
Antibody-sandwich ELISAs may be the most useful of the immunosorbent assays for detecting antigen because they are frequently between 2 and 5 times more sensitive than those in which antigen is directly bound to the solid phase. To detect antigen, the wells of microtiter plates are coated with specific (capture) antibody followed by incubation with test solutions containing antigen. Unbound antigen is washed out and an antigen-specific antibody conjugated to enzyme (i.e., developing reagent) is added, followed by another incubation. Enzyme labeled antibody can be produced in the same animal that produced passively adsorbed antibody, or from a different species immunized with the same antigen that is captured. Unbound conjugate is washed out and substrate is added. After another incubation, the degree of substrate hydrolysis is measured. The amount of substrate hydrolyzed is proportional to the amount of antigen in the test solution.
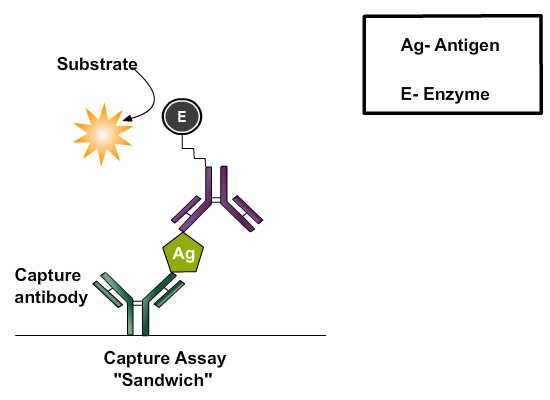
Different Stages Of Sandwich ELISA:
Coating ELISA Plates:
Coating is achieved through passive adsorption of the antibody to the assay microplate. This process occurs though hydrophobic interactions between the microtiter plate and non-polar protein residues. Although individual proteins may require specific conditions or pretreatment for optimal binding, the most common method for coating plates involves adding a 2-10 μg/ml solution of protein dissolved in an alkaline buffer such as phosphate-buffered saline (pH 7.4) or carbonate-bicarbonate buffer (pH 9.4).The buffer contains no other proteins that might compete with the target antigen for attachment to the microtiter plate. Antigens, which are protein in nature will attach passively to the microtiter well plate during incubation in incubator at 370C.
Washing step:
After incubation any excess antibody is removed by washing steps by flooding and emptying the wells with neutral phosphate buffered saline ( PBS ) or deionized water. Washing steps are necessary to remove nonbound reagents and decrease background, thereby increasing the signal to noise ratio. Insufficient washing will allow high background, while excessive washing might result in decreased sensitivity caused by elution of the antigen from the well.
Add blocking buffer:
The binding capacity of microplate wells is typically higher than the amount of protein coated in each well and the residual binding capacity of the plate is blocked in this step. The ideal blocking buffer will bind to all potential sites of nonspecific interaction, eliminating background altogether, without altering or obscuring the epitope for antibody binding. The blocking buffer is effective if it improves the sensitivity of an assay by reducing background signal and improving the signal-to-noise ratio. Tween 20 (0.05%) by itself is more effective at blocking than any protein tested, but because the combination of protein and Tween 20 may be more effective than Tween 20 alone in some cases, bovine serum albumin (BSA; 0.25%) is included in the blocking buffer.
Coated plates can be used immediately or dried and stored at 4°C for later use, depending on the stability of the coated protein.
Add antigen solution:
This step involves the addition of detecting soluble antigens (in test sample) being directed against the coated antibody. The antigen is usually diluted in blocking buffer to prevent non specific attachment of protein in the antiserum on the solid phase. The antibody present in the serum which are specific to the antigen, binds the coated antigen on incubation.
Washing step:
Excess antigen or unbound antigen are removed by washing step and is followed by addition of blocking solution.
Add antibody enzyme conjugate:
The next step is the addition of antibody enzyme conjugate, diluted in blocking buffer directed against the antigen. The choice of antibody enzyme conjugate is determined by the goals of the assay. If it is necessary to detect all antibodies that bind to antigen, conjugates prepared with antibodies specific for Ig κ and λ light chains should be used. Alternatively, protein A or protein G–enzyme conjugates may be preferable when screening monoclonal antibodies. Such antibodies are produced against immunoglobulins(Igs) of species in which the detecting antibodies are produced and are termed antispecies conjugates. Thus, if detecting antibodies are produced in rabbits, the enzyme-labeled antibodies would have to be antirabbit Igs in nature. This allow greater flexibility in use of antispecies conjugates in that different specificities of conjugate can be used to detect particular Igs binding in the assay. For example, the antispecies conjugate could be anti- IgM, IgG1, IgG2 and so on. The enzyme can be linked to a protein such as streptavidin if the primary antibody is biotin labeled. The most commonly used enzyme labels horseradish peroxidase (HRP) and alkaline phosphatase (AP). Other enzymes have been used as well, but they have not gained widespread acceptance because of limited substrate options. These include β-galactosidase, acetylcholinesterase and catalase.
Washing step: Unbound antibody enzyme conjugate is washed away after incubation phase.
Adding substrate:
Substrates are critical for the detection and visualization steps of an ELISA. The step involves the addition of suitable substrate solution for the particular enzyme conjugated to the antibodies. The objective is to allow development of color reaction through enzyme catalysis. A large selection of substrates is available for performing the ELISA with an HRP or AP conjugate. TMB (3, 3’, 5, 5’-tetramethyl benzidine) is the most commonly used substrate for the enzyme horseradish peroxidase (HRP). The substrates of alkaline phosphatase (AP) ,4-methylumbelliferyl phosphate (MUP) and pNPP (p-Nitrophenyl-phosphate) are nontoxic and relatively stable. Solutions of p-nitro-phenyl phosphate (NPP) are stable for months at 4°C, while solutions of 4-methylumbelliferyl phosphate (MUP) can be kept for months at room temperature without any significant spontaneous hydrolysis. The biggest disadvantage if NPP is used as a substrate is that, the yellow color of the nitrophenyl product is relatively difficult to detect visually. Using the substrate MUP instead of NPP can greatly enhance the sensitivity of the assay. The fluorogenic system using MUP is 10 to 100 times faster than the chromogenic system using NPP, and appears to be as sensitive as an enhanced chromogenic assay in which alkaline phosphatase generates NAD+ from NADP. The disadvantage of using fluorogenic substrates is that they require a microplate fluorometer costing twice as much as a high quality microtiter plate spectrophotometer.
The choice of substrate depends upon the required assay sensitivity and the instrumentation available for signal-detection (spectrophotometer, fluorometer or luminometer)
Stop solution:
The reaction is allowed to progress for a defined period after which the reaction is stopped by altering the pH of the system. Stop Solution is a used to terminate the enzyme substrate reaction for ELISA applications after attaining the desired color intensity which is an indication of analyte level. For eg. The TMB substrate reacts with immobilized horseradish peroxidase (HRP) conjugated antibodies to produce a blue solution. Reaction may be stopped by 0.2 M sulphuric acid which offers a yellow end product read at 450 nm. AP stop solution (0.5M NaOH) does not change the yellow color or the absorbance of the chromogen, and so the absorbance is read at 405 nm to 420 nm.
Quantification:
Specially designed spectrophotometers are available which reads through the microtiter wells either singly or in rows. Several ELISA plate readers are available, with increasing levels of sophistication. Some of these provide a measurement of optical density while some tabulate data and apply statistical analysis. Compatibility with a small computer, and availability of a suitable program to process the results and transform the optical density readings into concentrations of protein are important additional things to look for when selecting an instrument. Most ELISA readers can be set to measure the absorbance of the colors produced by the action of antibody- conjugated enzymes on their respective substrates. The microplate reader works by shining a particular type of light at each of the samples in the microwell plate. Common detection modes for microplate assays are absorbance, fluorescence intensity, luminescence, time-resolved fluorescence and fluorescence polarization. A light source illuminates the sample using a specific wavelength (selected by an optical filter, or a monochromator), and a light detector located on the other side of the well measures how much of the initial (100%) light is transmitted through the sample, the amount of transmitted light will typically be related to the concentration of the molecule of interest. This is called absorbtion detection. The range of application of fluorescence intensity detection is much broader than when using absorbance detection, but instrumentation is usually more expensive. Microplate readers feed the absorbance or fluorescence measures into a computer program that analyses the particular information being collected.
Assay optimization:
Serial dilution titration analyses are performed to determine optimal concentrations of reagents to be used in ELISAs. All three reactants in ELISA, a solid-phase coating reagent, a secondary reagent that binds the primary reagent, and an enzyme-conjugated tertiary developing reagent that binds to the secondary reagent are serially diluted and analyzed by a criss-cross matrix analysis. Once the optimal concentrations of reagents to be used under particular assay conditions are determined, these variables are kept constant from experiment to experiment.
Assay validation:
ELISA kits that are commercially available which are used for diagnostic purposes in the detection of specific antigen or antibody in the serum sample. For eg ,ovarian cancer antigen (CA-125) enzyme immunoassay test kit is intended for use as a monitoring and screening test for serum CA-125 level. An elevated serum CA-125 level can indicate ovarian cancer and suggests the need for further clinical management, also determining serum CA-125 concentration may be useful in monitoring patients with diagnosed ovarian cancer. Materials provided with the test kits includes antibody coated microtiter plate with 96 wells, enzyme conjugate reagent, substrate solution, stop solution, wash buffer concentrate, sample diluent, reference standards, positive and negative controls.
ELISA results are reported as a number and the most controversial aspect of this test is determining the "cut-off" point between a positive and negative result. A cut-off point may be determined by comparing the ELISA plate reader value with a known reference standard. If an ELISA test is used for drug screening at workplace, a cut-off concentration, 50 ng/mL, for example, is established, and a sample will be prepared which contains the standard concentration of analyte. Unknowns that generate a signal that is stronger than the known sample are "positive" and those that generate weaker signal are "negative."